

AtlasXomics Data Analysis Package
Process raw data locally, on our cloud-based platform, or with a combination of the two.
Download our analysis scripts and tutorials and process the data in your HPC or local environment
Upload your raw data to AtlasXomics’ cloud-based environment for automated analysis

Raw data generated by the AtlasXomics platform
DBiT-seq combines microfluidics and Next-Generation Sequencing (NGS) to generate high-quality spatial data from a wide range of tissues. The assay produces microscopy and sequencing data for each sample.

-
The workflow starts by importing microscope images into AtlasXBrowser, our custom app for DBiT-seq image processing. This is done to identify on-tissue and off-tissue areas, and to produce the tissue position file which locates the spatial barcodes on tissue. Following this, the raw sequencing data undergoes processing through our custom epigenomic preprocessing and alignment workflow. This results in the generation of fragment files organized by pixel barcode. Once processed, the data can be inputted into ArchR or Signac, and then visualized in Seurat. Alternatively, the data can be further refined using our automated workflows available in our cloud-based workspace. Within this cloud environment, samples are combined and filtered. Cluster optimization is performed. Selected parameters are subsequently used to analyze all samples in a project, ultimately yielding differential epigenomic elements organized by clusters and samples. This data can be downloaded as processed Seurat and ArchR projects or can be viewed in our tailored Shiny app.

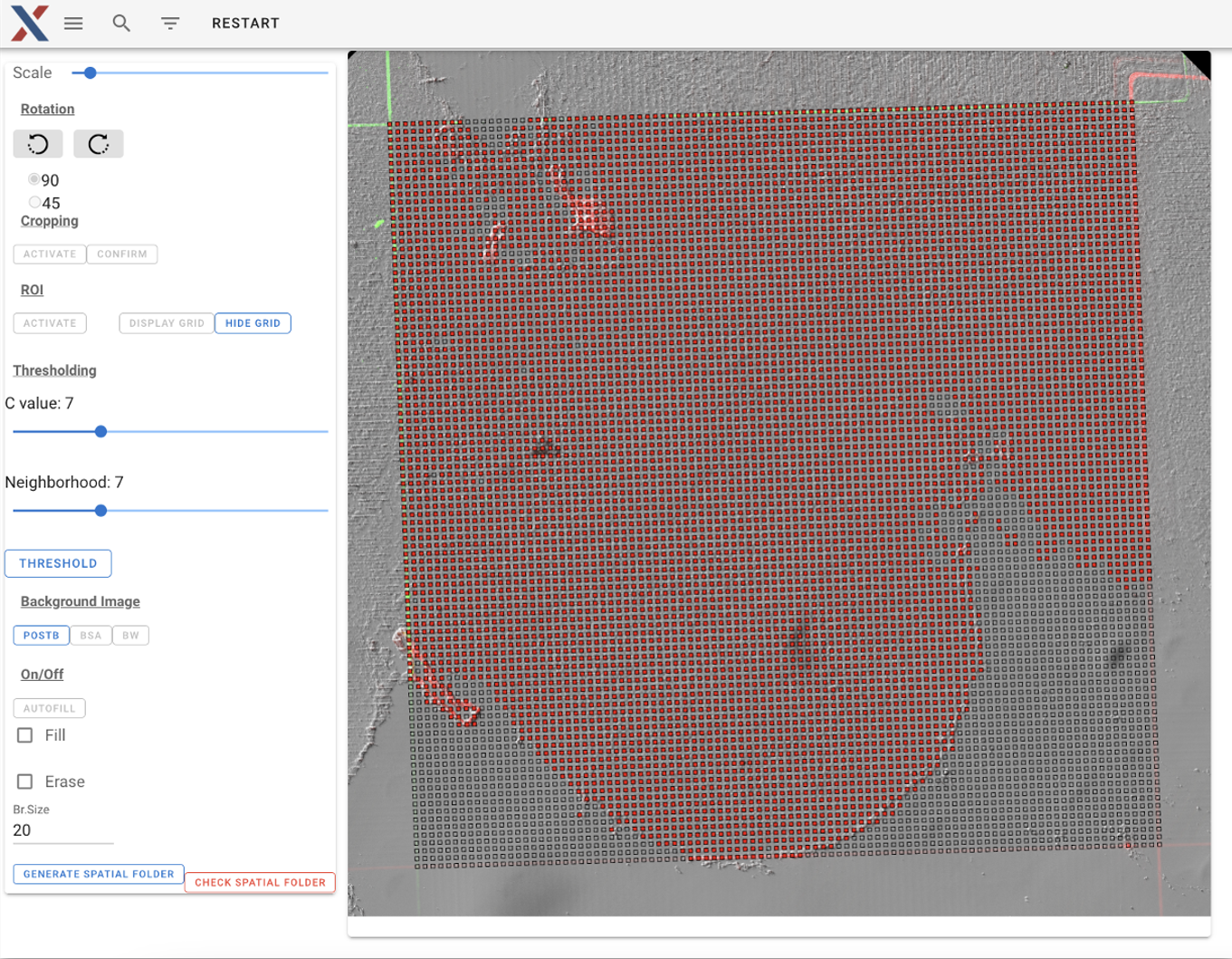
Step 1: Generate position file with AtlasXBrowser
Load post-microfluidics images into AtlasXBrowser either locally or in the Latch workspace. Identify on/off tissue pixels with a combination of automatic calling and manual selection.
Threshold image for automatic on/off tissue calling

Manually select on tissue regions (red boxes)
Step 2: Process sequencing data and align to genome
FASTQ files can be processed on our cloud platform or locally with our Snakemake workflow. The process involves the following: reads are filtered on the correctness of ligation linker sequences; alignment and preprocessing is performed with Chromap; BED output files are converted into fragment.tsv.gz files for downstream analysis.

Transfer or download processed raw data for further analysis in R
Processed raw data (fragment and position files) can be downloaded and analyzed locally. Data is compatible with many open-source packages including ArchR and Signac to identify clusters and differential epigenetic elements. Calculated clusters, motifs, and gene activity scores can be loaded into Seurat with the position files from Step 1 to visualize spatially.
See the ATX_epigenomics GitHub for a downstream processing tutorial.
Alternatively, the data can be processed in our cloud platform (Step 3).

Step 3: Select best clustering parameters and clean fragment file
Using the Optimize ArchR workflow, a wide range of clustering parameters can be visualized to determine the optimal set. In addition, using the Cleaning workflow, row and column artifacts can be minimized or removed prior to analysis

Step 4: Generate spatial epigenome data with selected parameters
Using one of our workflows, differential genes, peaks, and motifs by condition, cluster, and sample are outputted with fully processed ArchR and Seurat objects. Data can be downloaded or loaded into our custom Shiny app for visualization.

Step 5: Visualize processed data in our interactive shinyApp
Load processed data into our interactive browser to easily explore spatially differentiated epigenetic elements in tissue.
